|
For the PC owners: Have you ever used Windows PowerShell? It's something akin to Command Prompt, and can be utilized for task automation. It provides a really easy way to batch rename hundreds of files in a folder. Specifically, I had hundreds of individual file names containing a "-" that needed to be replaced or removed, and I was looking for something straightforward for changing the names. I found Windows PowerShell, which is a management framework that was developed about ten years ago. I discovered it was pretty easy to use PowerShell for this task and others like it, and thought I'd share it here! Here's how to get set up: All your files should be in one folder, which you'll designate as your working directory. Open PowerShell on your PC. You can just type "powershell" into the search box in Windows 10. A window will open. To navigate to your folder of files, type "cd" (change directory) at the prompt and then add a space. Drag and drop your working folder into the PowerShell window. The drag and drop puts the whole file path into place for you. Press enter and you'll see that you're now operating out of your folder. 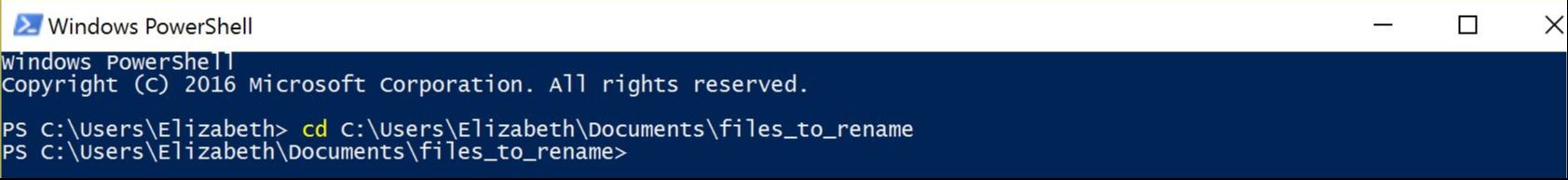 My files are all in a folder called "files_to_rename". This folder is now set as my working directory. to do a batch rename on the files in your current directory:
How does it work? You are 'piping' ("|") the contents of your directory ("dir") to the "rename-item" cmdlet (read: command-lette). If you were to enter only "dir" at the prompt, you would see all the files in your folder displayed; we use the last section of the script to run through all of these files. I initially learned of this command at: https://www.howtogeek.com/111859/how-to-batch-rename-files-in-windows-4-ways-to-rename-multiple-files/
0 Comments
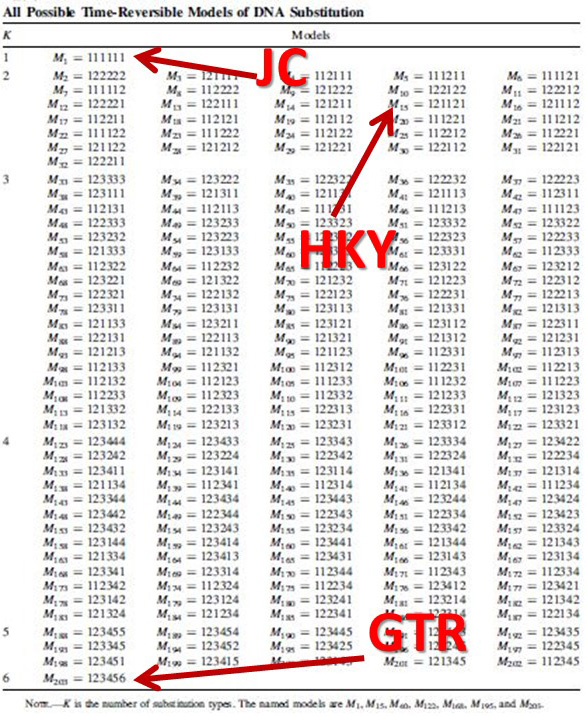
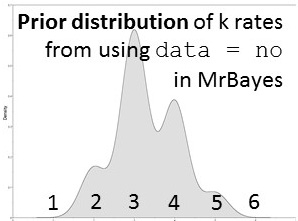
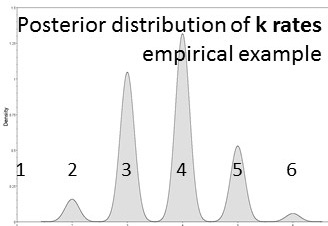
nst=mixed. This is one of my favorite commands in MrBayes. It's used instead of designating a nucleotide substitution model a priori. Instead, reversible jump is a form of model-averaging (across different dimensions of parameter space) where all possible time-reversible substitution models are explored, incorporating uncertainty in model selection.
Likelihood-based analyses results are dependent on the model-fit to the data. The credible set of substitution models typically contains several different models and the benefit of rjMCMC is that it allows integration of all. Though tree topology isn't extremely sensitive to model misspecification, other parameters may be (Alfaro & Huelsenbeck 2006). example of mrbayes block using rjMCMC: [after the nexus alignment of data, use these commands for rjMCMC in MrBayes; this shows data grouped in two subsets] BEGIN MRBAYES; log start filename=murray-rj_log; charset codon_pos1 = 1-1041\3; charset codon_pos2 = 2-1041\3; charset codon_pos3 = 3-1041\3; partition two_subsets = 2: codon_pos1 codon_pos2, codon_pos3; set partition = two_subsets; lset applyto=(all) nst=mixed rates=gamma; [each of the two subsets will be treated separately] [use 'nst= mixed' for rjMCMC and 'rates=gamma' to incorporate rate heterogeneity; I don't use parameter for invariant sites] unlink shape=(all) pinvar=(all) statefreq=(all) revmat=(all); prset applyto=(all) ratepr=variable; mcmc ngen=1000000 samplefreq=100 filename=murray_rj; sumt; sump; [gives a default 25% burnin for tree and parameter files] log stop; end; Also a great tool -- rjMCMC in BEAST 2! Just install the RBS plugin while in BEAUti and you are set to go. The drawback -- as of now (Dec. 2016) you cannot run the XML file in CIPRES if you are using reversible jump models from the RBS plugin. note:
references:
Huelsenbeck, J.P., Larget, B. & Alfaro, M.E. (2004) Bayesian phylogenetic model selection using reversible jump Markov chain Monte Carlo. Mol Biol Evol, 21, 1123-33. Alfaro, M.E., & Huelsenbeck, J.P. (2006). Comparative performance of Bayesian and AIC-based measures of phylogenetic model uncertainty. Syst Biol, 55, 89-96. |
PhyloBlogCovering topics of phylogenetics and systematics & other science-related news. Archives
October 2019
Categories
All
|
 RSS Feed
RSS Feed