|
When deciding how to start out the first day of the semester of my insect diversity and evolution course, I devised a quick activity to get the students interacting over insects.
For the first lecture, I had the students grab a unit tray as they arrived -- each tray was placed upside-down in the drawer. The insects themselves weren't anything special (though that could be a fun twist), but they served as a group-forming tool to get students interacting.
At the very beginning of class, students had to walk around and find the three other people with the same insect, and then they each spent a minute or two introducing themselves to the group. It is a more personal setting than immediately going around the room for roll call and introductions. Sometimes class-wide student introductions can be a little nerve-racking on the first day, so I thought it would be nicer to start in small groups before taking attendance.
1 Comment
Setting a starting tree in the program BEAST can be a complicated issue, and I've been asked about troubleshooting for it. Here is a full XML file with annotations, as an example of how to designate a starting tree and how to force BEAST to keep it as a fixed topology. BEAST is a widely-used phylogenetic dating program and has an excellent GUI interface in BEAUti, where users can control most all parameters and inputs they'd need. BEAUti is the front-end program that produces the XML file that is then used by BEAST for tree estimation. XML stands for 'eXtensible Markup Language', which is both human and machine readable, and is similar to HTML. One piece that must be manually edited in a text editor is the user-specified starting tree, if desired. Why use a starting tree? For large and difficult datasets, one can start in the best area of parameter space, so that the Markov chain isn't wasting time jumping around to sample the presumably 'incorrect' topologies. I'm not sure as to how much a starting tree increases efficiency, since alternate topologies can still be sampled (it's certainly not needed for small, straightforward datasets), but I may update my opinions in the future based on the success of trying to manipulate phylogenomic data. There are two different XML editing tasks I'll cover.
Setting a user-specified starting topology The default starting tree is a random tree, which is coded in an element (content & attributes surrounded by an opening and closing tag ('init', our tree initializer). The whole element needs to be replaced by a user-specified starting tree in newick format. Take a look at the example document! Fixing the starting tree topology There are four lines to comment out: the operators for subtree-slide, wide & narrow exchange, and Wilson-Balding. Removing these four operators prevents the topology from updating, but still allows for estimation of the node ages (i.e., branch lengths will be modified even though the topology will not). In an XML file, comments are surrounded by ' <!-- ' and ' --> ', which means they will not be processed.
There are a couple of nice sites with information on user input starting trees, yet translating that to your own data can still be a bit of a struggle. I hope here I could add a bit of guidance on the issue by providing an annotated XML file to help clarify the changes needed.
There is now an updated and more-detailed tutorial at: http://www.beast2.org/fix-starting-tree/. Also, thanks to great info from: http://www.northernbotanist.com/?page_id=732. KUCR -- call letters for the University of California Riverside's award winning radio station. My friend Dr. Tony Yang interviewed me on his radio show, I'll Look Into It, and we talked about my research on wasps & discussed why biologists study evolutionary relationships.
About the radio show: I’ll look Into It "is appointment listening for individuals looking for a higher level of discussion. ... The show features a Science Today segment that features experts in scientific fields that will soon impact our daily lives, and the difficult basic science research done to achieve breakthroughs."
In the interview, we talked mainly about wasps and phylogenies. I introduced the fact there there are two different groups of wasps, generally. The parasitoids and the more familiar predators, such as the stinging paper wasps and hornets. Parasitoids are tiny and important for biological control, which is a term first used at the Mission Inn in downtown Riverside back in 1919! The radio interview is less than a half hour. It was my first one, which was a bit of a learning experience, and it was great to have done it -- I really appreciate that Tony thought of me and my work and wanted to feature me on his show! The EvoGroup at Cornell recently hosted the annual EvoDay conference, held at the Lab of Ornithology. This year's theme was 'Phylogenomics'.
Each year at EvoDay there is a mix of faculty, researchers, postdocs, and grad students who give ~20 minute presentations at the event, addressing an audience of about 100 people. Past themes have been 'Evolution and Conservation' (2016) and 'Evolution and Behavior' (2015).
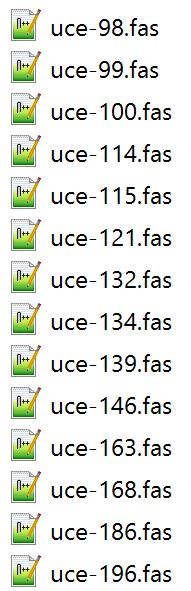
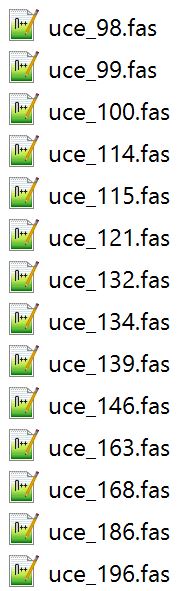
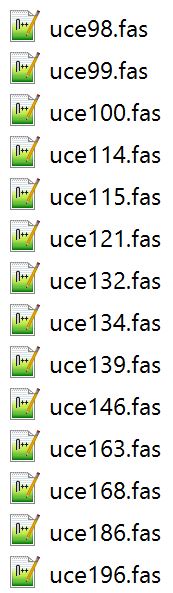
This year, our aculeate working group was well-represented, as both Bonnie Blaimer (Smithsonian) and I presented our latest work based on phylogenies constructed from ultraconserved elements (UCEs). For the PC owners: Have you ever used Windows PowerShell? It's something akin to Command Prompt, and can be utilized for task automation. It provides a really easy way to batch rename hundreds of files in a folder. Specifically, I had hundreds of individual file names containing a "-" that needed to be replaced or removed, and I was looking for something straightforward for changing the names. I found Windows PowerShell, which is a management framework that was developed about ten years ago. I discovered it was pretty easy to use PowerShell for this task and others like it, and thought I'd share it here! Here's how to get set up: All your files should be in one folder, which you'll designate as your working directory. Open PowerShell on your PC. You can just type "powershell" into the search box in Windows 10. A window will open. To navigate to your folder of files, type "cd" (change directory) at the prompt and then add a space. Drag and drop your working folder into the PowerShell window. The drag and drop puts the whole file path into place for you. Press enter and you'll see that you're now operating out of your folder. 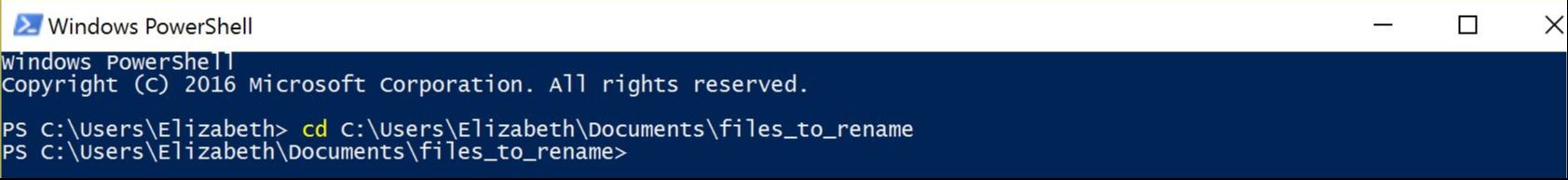 My files are all in a folder called "files_to_rename". This folder is now set as my working directory. to do a batch rename on the files in your current directory:
How does it work? You are 'piping' ("|") the contents of your directory ("dir") to the "rename-item" cmdlet (read: command-lette). If you were to enter only "dir" at the prompt, you would see all the files in your folder displayed; we use the last section of the script to run through all of these files. I initially learned of this command at: https://www.howtogeek.com/111859/how-to-batch-rename-files-in-windows-4-ways-to-rename-multiple-files/
|
PhyloBlogCovering topics of phylogenetics and systematics & other science-related news. Archives
October 2019
Categories
All
|
||||||||||||||||


 RSS Feed
RSS Feed